一针 2300 万、曾刷新世界最贵药物榜单的疗法 Elevidys,最近接连报告死亡病例。
上周五,美国 FDA 出于安全考虑,要求这一疗法停售。然而,药企却强硬地表示拒绝,公开与 FDA 对抗。
到底发生了什么?
世界最贵疗法之一,接连出现 2 例死亡
要聊清楚 Elevidys 身上的争议,我们先把时间倒回 2 年前。
2023 年 6 月 22 日,一款名为 Elevidys 的药物获得 FDA 加速批准。随后,这款定价为 320 万美元一个疗程的新药迅速跻身上榜,成为了当前全世界第二贵的药。(点击查看丁香园往期内容:2300 万一针!刷新世界最贵药榜单,但不一定能挣到钱……)
该药由 Sarepta 和罗氏共同开发,是全球首款杜氏肌营养不良(DMD)基因治疗药物。
DMD 是一种罕见的遗传性肌肉疾病,婴幼儿发病率 1/3300,且致残率极高,典型的患者早在 2~3 岁时就会发病,12~13 岁时就只能依赖轮椅生活,平均活不过 20 岁 [1]。
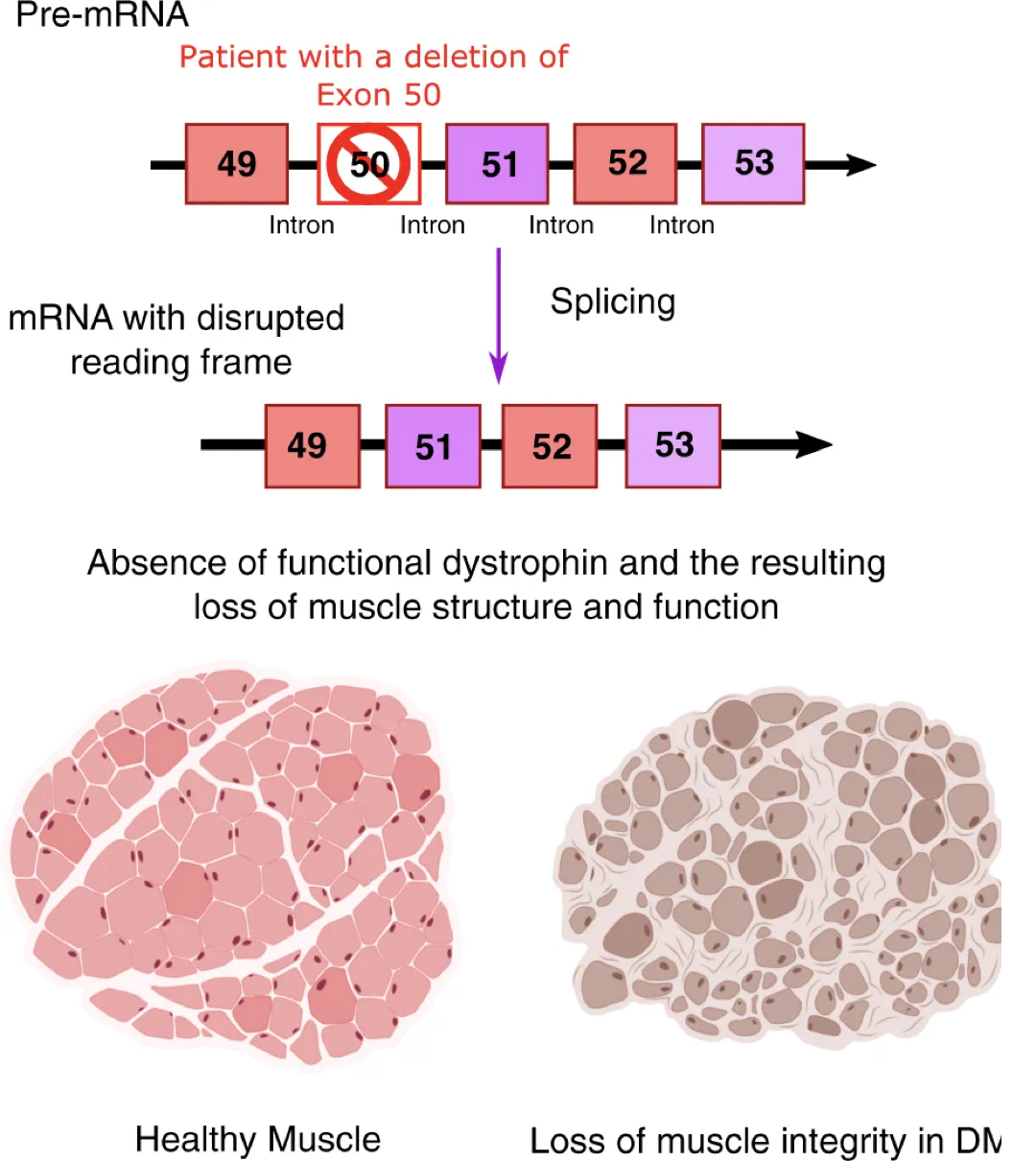
抗肌萎缩蛋白基因突变导致患者难以形成正常肌肉(图源:参考资料 2)
作为一种遗传病,一种理想的治疗思路是:将正常的抗肌萎缩蛋白基因导入肌纤维,患者就有可能恢复正常的肌肉功能。
但是,抗肌萎缩蛋白基因是目前所知最大的人类基因,常用的一些基因治疗载体无法携带完整的基因,这也让 DMD 的基因治疗研发受到广泛关注。
在这其中,最亮眼的就是 Sarepta 的 Elevidys。2019 年,跨国巨头罗氏制药为之直接注资 11.5 亿美元(约合 83 亿人民币),还外加最高 17 亿美元里程碑付款 [3]。
终于,在经过数年研发后,Elevidys 以当时全球第二贵的高价问世,获得 FDA 加速批准用于治疗 4~5 岁患有 DMD 且已确认 DMD 基因发生突变的儿童患者。

FDA 对 Elevidys 的审批文件截图
2024 年 6 月,这项加速批准正式转为全面批准,同时 Elevidys 还获得了新的加速批准,适用人群年龄扩大到 4 岁及以上。
在当时,FDA 生物制品评估与研究中心主任 Peter Marks 医学博士表示:「今天的批准扩大了适合接受这种疗法的杜氏肌营养不良症患者范围,有助于解决这种毁灭性且危及生命的疾病患者持续而迫切的治疗需求。 」
但很快,问题出现了。
2025 年 3 月 18 日,Elevidys 报告出现首例死亡病例。
根据 Sarepta 发表声明,死亡患者是一名患有非行走型杜氏肌营养不良症的年轻人,死因为急性肝衰竭。
经进一步检测发现,这名患者近期曾感染巨细胞病毒(CMV),主治医生认为这可能是导致此次事件的促发因素——CMV 可感染肝脏并引起损伤,这种情况也被称为 CMV 肝炎。
Sarepta 表示,急性肝损伤是 Elevidys 及其他腺相关病毒(AAV)介导的基因疗法已知的潜在副作用之一,这一点已在 Elevidys 处方标签中予以明确说明。
然而,不到 3 个月,Elevidys 再次出现第二例死亡病例。
6 月 15 日,Sarepta 公告表示,另一名非行走型 DMD 年轻患者在接受临床试验用药后死亡,死因同样为急性肝衰竭。随后,Sarepta 表示自愿暂停一项研究,FDA 表示已介入调查。
而就在上周四,Sarepta 再次披露了一例死亡。这次,患者使用的并非 Elevidys,而是另外一款名为 SRP-9004 的在研基因疗法(处于 I 期临床试验中)。该公司向媒体证实,死亡原因同样与肝毒性有关。
曾在争议下获批,适应症逐步扩大
实际上,Elevidys 面临的争议,从两年前获批开始就一直存在——它是史上唯一一个没有硬性疗效证据,而是通过 FDA 加速审评程序上市的基因治疗药物。
在 Nature 同期发布的报道,标题上甚至赫然写着「内部反对」。
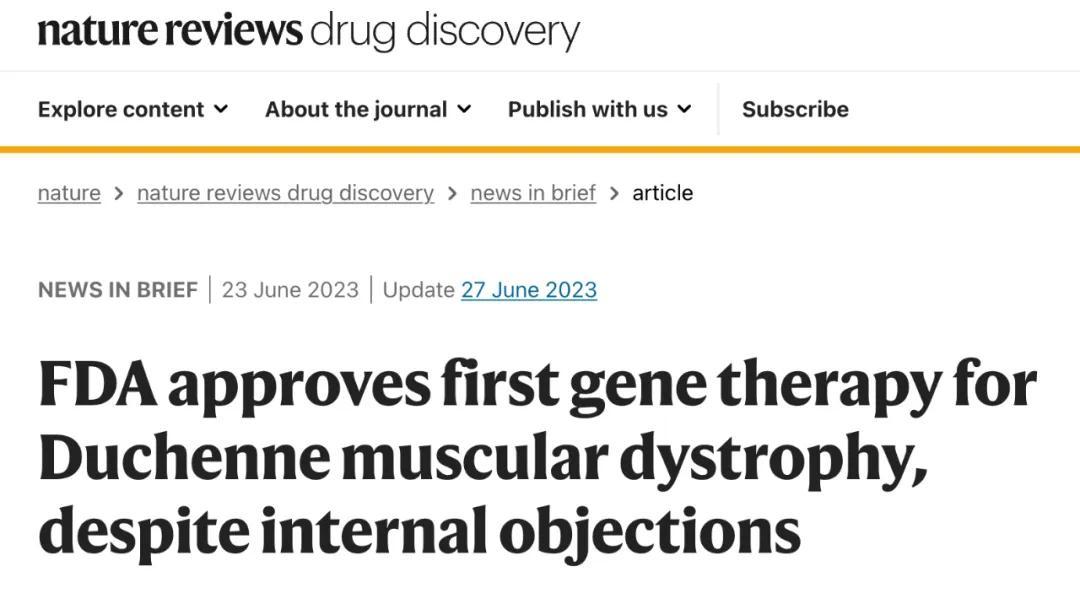
Nature 报道将「内部反对」写进了标题(图源:参考资料 4)
在 FDA 关于该药上市的专家咨询委员会会议上,该药的专家投票是 8 票赞成、6 票反对[5]。
最大的问题是,审批依据的临床试验并未直接设置疗效终点,而是用了「患者骨骼肌中观察到了截短型抗肌萎缩蛋白的表达」这个替代终点。
反对的专家们认为,这只能说明 Elevidys 成功将基因导入了肌纤维中并稳定表达,无法证明其是否与患者的临床症状的改善是否有关。
在 II 期临床试验中,治疗组患者第 12 周的肌肉活检显示,肌纤维抗肌萎缩蛋白水平已经达到了正常人的 40% 左右;但在第 48 周的行走能力评价量表中,治疗组和安慰剂组的分数则并无差异。
进一步分析则显示,Elevidys 成功提高了 4~5 岁儿童的行走能力,但对 6~7 岁儿童则并无效果。也正因如此,FDA 的审批才加上了 4~5 岁这样一个严格的年龄限制 [6]。
另一方面,虽然是天价药,但大多数人对其未来的市场表现也不看好。在获得 FDA 加速批准后,多数报道认为,Elevidys 疗效轻微且适用患者非常少,因此很难盈利 [8]。
原因很简单:2300 万一个疗程,太贵了;本来就是罕见病,还严格限制 4~5 岁,能用的患者太少了。
就如同曾经上榜「全球十大最贵药物」的 Beqvez、Skysona 和 Zynteglo,刚获批时风光无两,但短短几年间就纷纷因「企业战略问题」相继在全球或欧洲区退市。(点击查看丁香园往期内容:一针 2500 万,能买北京四合院!全球最贵的药,才卖一年就退市了…)
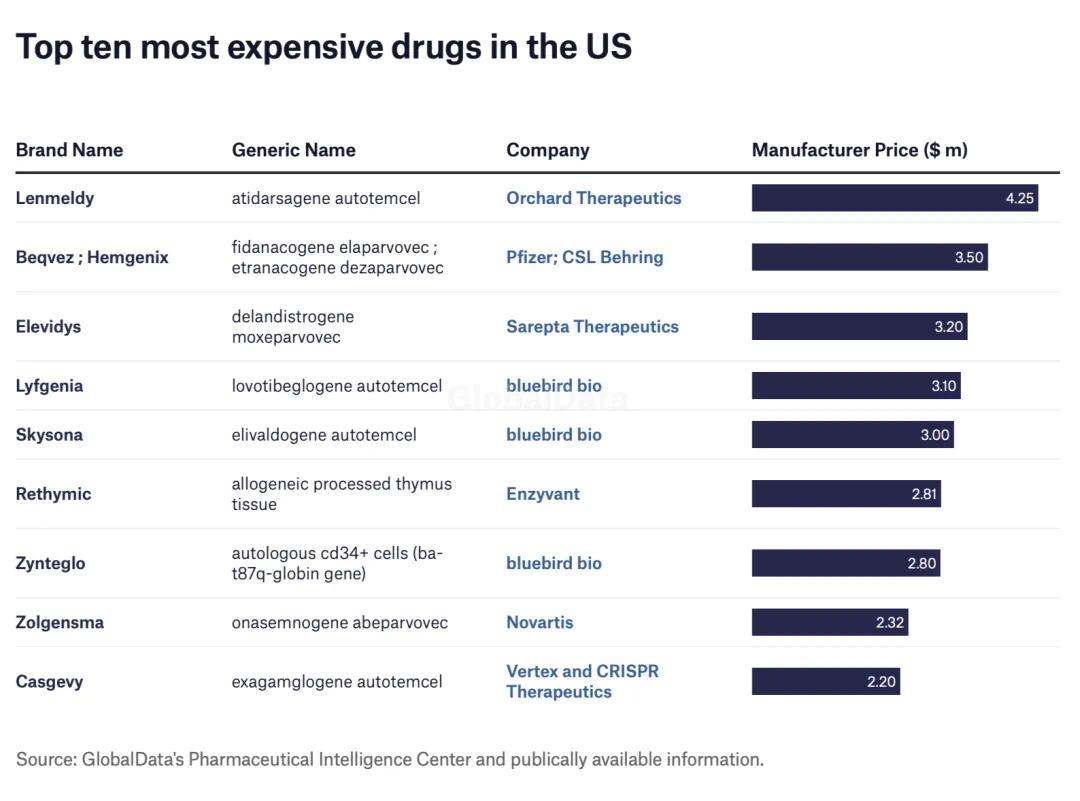
图源:参考资料 7
为了避免走上这条老路,Elevidys 选择扩大适应症。
根据两项临床研究的结果,Elevidys 于 2024 年 6 月获得 FDA 批准大幅扩展适应症,从原来的仅限于 4~5 岁具有行走能力的儿童,扩展为 4 岁及以上的儿童(无论是否有行走能力)[9]。
根据 Sarepta 2024 财报,Elevidys 上市后的第一年,全年净收入 8.208 亿美元,总额已进入「10 亿美元俱乐部」,预计 2025 年销售额能达到 30 亿美元左右。
裁员 36%,公司还要硬刚 FDA:拒绝停售
然而,接连两例死亡报告,为 Elevidys 的前途投下了一片阴影。
公布第二例死亡病例的同时,Sarepta 宣布,为加强非行走型 DMD 患者安全性,已自愿停止为非行走型的 DMD 患者提供 Elevidys,同时自愿暂停一项临床研究。
当地时间 7 月 16 日,Sarepta 发表声明表示,已依据 FDA 要求为 Elevidys 补充针对急性肝损伤和急性肝衰竭的黑框警告。[10]

Sarepta 官网截图
这份声明还提到,公司计划裁员约 36%,并暂停多个药物的研究,以节约成本应对危机。
当地时间 7 月 18 日,FDA 宣布要求 Sarepta 暂停所有 Elevidys 的销售,以及多种基因疗法的临床试验[11]。有 FDA 官员在接受采访时表示,「正在认真考虑将该疗法从市场上撤回」[12]。

FDA 官网截图
FDA 生物制品评估与研究中心主任表示:「保护患者安全是我们的首要任务,FDA 不会批准任何弊大于利的产品。如果临床试验参与者面临不合理且重大的患病或损伤风险,FDA 将暂停任何临床试验。」[11]
然而,Sarepta 却在与 FDA 领导层的会面中直接拒绝了停售要求[12]。
Sarepta 在 7 月 18 日发布的最新声明[13]中表示:「基于我们对数据的全面科学解读,在行走型患者群体中,没有出现新的安全信号,我们将继续向行走型患者群体供应 Elevidys。」
策划:z_popeye|监制:islay
题图来源:视频截图
参考资料:
[1]Passamano L, Taglia A, Palladino A, et al. Improvement of survival in Duchenne Muscular Dystrophy: retrospective analysis of 835 patients. Acta Myol. 2012;31(2):121-5.
[2]Himič V, Davies KE. Evaluating the potential of novel genetic approaches for the treatment of Duchenne muscular dystrophy. Eur J Hum Genet. 2021;29(9):1369-1376. doi: 10.1038/s41431-021-00811-2
[3]F. Hoffmann-La Roche Ltd. Roche enters licensing agreement with Sarepta Therapeutics to improve the lives of patients living with Duchenne muscular dystrophy. https://www.roche.com/media/releases/med-cor-2019-12-23
[4]U.S. Food and Drug Administration. FDA Approves First Gene Therapy for Treatment of Certain Patients with Duchenne Muscular Dystrophy.
[5]Mullard A. FDA approves first gene therapy for Duchenne muscular dystrophy, despite internal objections. Nat Rev Drug Discov. 2023 Jun 23. doi: 10.1038/d41573-023-00103-y
[6]BioSpace. Sarepta Shares Fall Due to Concerns Over Elevidys’ Potential for Label Expansion. https://www.biospace.com/article/fda-approves-sarepta-s-gene-therapy-for-duchenne-muscular-dystrophy
[7]Sabatini MT, Chalmers M. The Cost of Biotech Innovation: Exploring Research and Development Costs of Cell and Gene Therapies. Pharmaceut Med. 2023;37(5):365-375. doi: 10.1007/s40290-023-00480-0
[6]https://www.fiercepharma.com/pharma/pfizer-scores-fda-nod-hemophilia-b-gene-therapy-will-charge-35m-dose
[7]https://link.springer.com/article/10.1007/s40290-023-00480-0
[8]Mullard A. FDA approves first gene therapy for Duchenne muscular dystrophy, despite internal objections. Nat Rev Drug Discov. 2023 Jun 23. doi: 10.1038/d41573-023-00103-y
[9]https://www.fda.gov/news-events/press-announcements/fda-expands-approval-gene-therapy-patients-duchenne-muscular-dystrophy
[10]https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-announces-strategic-restructuring-and?_ga=2.34941473.160626044.1753073361-2101100171.1753073361
[11]https://www.fda.gov/news-events/press-announcements/fda-requests-sarepta-therapeutics-suspend-distribution-elevidys-and-places-clinical-trials-hold
[12]https://www.biopharmadive.com/news/sarepta-fda-elevidys-duchenne-stop-shipments-safety/753479/
[13]https://investorrelations.sarepta.com/news-releases/news-release-details/sarepta-therapeutics-provides-statement-elevidys?_ga=2.92737181.160626044.1753073361-2101100171.1753073361
编辑:ifhealth 来源:丁香园
